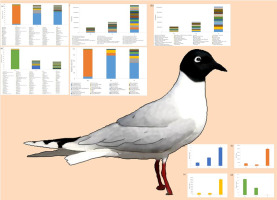
Gut microbiota and gene function prediction of Larus saundersi were analyzed.
•
Firmicutes (90.08%) was main phylum in AL, and Proteobacteria (77.67%) in YL.
•
Catellicoccus was main genus (88.99%) in AL, and Ralstonia (17.13%) in YL
•
Frequency of COG and KEGG pathways were both: S > YL > AL.
Abstract
This study explored gut microbiota and gene function prediction of young and adult Larus saundersi (YL, AL) in breeding season in Yellow River Delt. The main phylum was Firmicutes (90.08%) in AL, and Proteobacteria (77.67%) in YL, the main genus was Catellicoccus (88.99%) in AL, and Ralstonia (17.13%) in YL. AL and YL existed huge difference and bacterial composition in YL was of more similarities with its feeding ground (soil, S). Gene function prediction of AL and YL were enriched with cellular processes and signaling, metabolism and information storage and processing from clusters of orthologous groups (COG) pathways, AL and YL were enriched with environmental information processing, metabolism, genetic information processing from kyoto encyclopedia of genes and genomes (KEGG) pathways. This study would provide basic theoretical basis for epidemiology of migratory bird pathogenic microorganisms and intestinal microecology of migratory waterbirds.